

Khám phá thuốc nhanh hơn thông qua máy học
TNNN - Nghiên cứu kết hợp hóa học và máy học có thể giảm bớt các trở ngại trong quá trình phát hiện và sàng lọc thuốc.
- Máy học dự đoán mô hình thuốc tối ưu
- Máy học và cái nhìn toàn diện về các yếu tố gây ung thư vú
- Tìm thấy ở biển "thuốc giải độc" chất diệt cỏ paraquat
- Thuốc bepridil có tiềm năng điều trị coronavirus
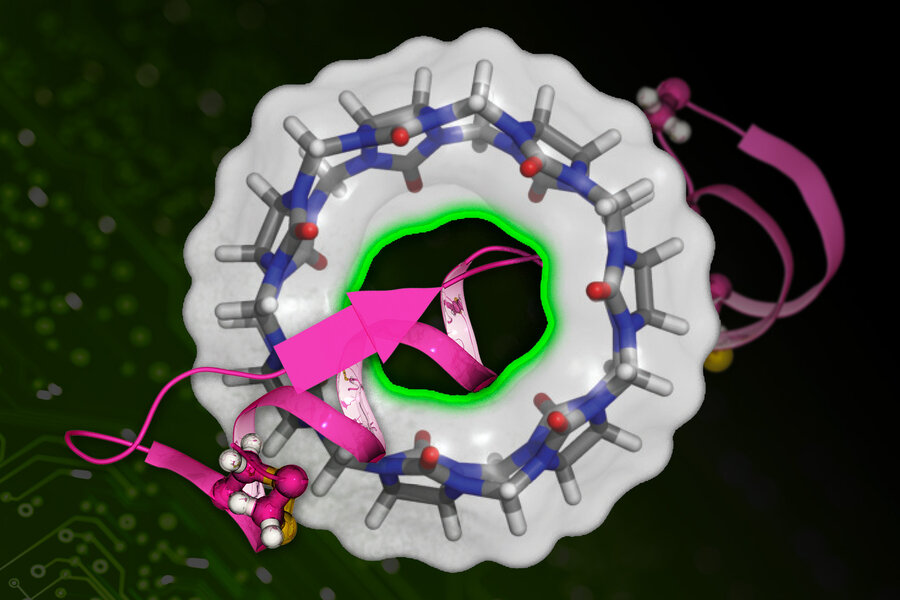
Các nhà nghiên cứu của MIT đã phát triển một kỹ thuật dựa trên máy học để tính toán nhanh hơn ái lực liên kết của một phân tử thuốc (được biểu thị bằng màu hồng) với một protein đích (cấu trúc hình tròn). Ảnh nguồn: MIT
Các loại thuốc chỉ có thể hoạt động nếu chúng dính vào các protein mục tiêu trong cơ thể. Các nhà nghiên cứu xác định, độ bám dính của thuốc là một trở ngại quan trọng trong quá trình phát hiện và sàng lọc thuốc. Giờ đây, nghiên cứu kết hợp hóa học và máy học mới có thể giảm bớt các trở ngại đó.
Kỹ thuật mới được đặt tên là DeepBAR sẽ nhanh chóng tính toán các ái lực liên kết giữa các loại thuốc thích hợp nhất và mục tiêu của chúng. Phương pháp này mang lại các phép tính chính xác và chỉ mất một phần nhỏ thời gian so với các phương pháp hiện đại trước đây. Các nhà nghiên cứu cho biết, một ngày nào đó DeepBAR có thể đẩy nhanh tốc độ khám phá thuốc và kỹ thuật protein.
Nghiên cứu được công bố trên Tạp chí Journal of Physical Chemistry Letters mới đây. Tác giả chính của nghiên cứu là Xinqiang Ding, một nghiên cứu sinh sau tiến sỹ tại Khoa Hóa học của MIT.
Bin Zhang, giáo sư tại MIT, thành viên liên kết của Viện Broad của MIT và Harvard, tác giả bài báo mô tả kỹ thuật này cho biết: “Phương pháp của chúng tôi có cấp độ nhanh hơn trước đây, nghĩa là chúng tôi có thể khám phá ra loại thuốc vừa hiệu quả vừa đáng tin cậy hơn”.
Ái lực giữa một phân tử thuốc và một protein nhắm đích được đo bằng một đại lượng gọi là năng lượng tự do liên kết - số lượng càng nhỏ, liên kết càng dính chặt.
Zhang nói: “Năng lượng tự do liên kết thấp hơn có nghĩa là thuốc có thể cạnh tranh tốt hơn với các phân tử khác, nó có thể phá vỡ chức năng bình thường của protein một cách hiệu quả hơn”.
Việc tính toán năng lượng tự do liên kết của một loại thuốc cung cấp một chỉ số về hiệu quả tiềm năng của một loại thuốc.
Các phương pháp tính toán năng lượng tự do liên kết được chia thành hai loại lớn, mỗi loại đều có ưu nhược điểm riêng. Một loại sẽ tính toán chính xác số lượng, lượng thời gian tiêu tốn và nguồn năng lượng máy tính. Loại thứ hai ít tốn kém hơn về mặt tính toán, nhưng nó chỉ cung cấp giá trị gần đúng của năng lượng tự do liên kết. Từ phương pháp này, Zhang và Ding đã nghĩ ra một cách tiếp cận để có được những điều tốt nhất của cả hai loại.
Chính xác và hiệu quả
DeepBAR tính toán chính xác năng lượng tự do liên kết, nhưng nó chỉ cần một phần nhỏ của các phép tính theo yêu cầu của các phương pháp trước đó. Kỹ thuật kết hợp mới này các phép tính hóa học truyền thống với những tiến bộ gần đây trong học máy.
"BAR" trong DeepBAR là viết tắt của "tỷ lệ chấp nhận Bennett", một thuật toán có từ hàng thập kỷ, được sử dụng để tính toán chính xác năng lượng tự do liên kết. Việc sử dụng tỷ lệ chấp nhận Bennet thường yêu cầu kiến thức về hai trạng thái "điểm cuối" (ví dụ: phân tử thuốc liên kết với protein và phân tử thuốc phân ly hoàn toàn khỏi protein), cộng với kiến thức về nhiều trạng thái trung gian (ví dụ: các mức độ liên kết một phần khác nhau), tất cả đều làm giảm tốc độ tính toán.
DeepBAR cắt giảm những trạng thái ở giữa đó bằng cách áp dụng tỷ lệ chấp nhận Bennett trong các khuôn khổ học máy được gọi là mô hình phát triển chuyên sâu. Zhang nói: “Những mô hình này tạo ra một trạng thái tham chiếu cho mỗi điểm cuối, trạng thái bị ràng buộc và trạng thái không liên kết”. Hai trạng thái tham chiếu này quá đủ để tỷ lệ chấp nhận Bennett có thể được sử dụng trực tiếp mà không cần tất cả các bước trung gian, rất tốn kém.
Khi sử dụng các mô hình tạo gen chuyên sâu, các nhà nghiên cứu đã tiếp cận lĩnh vực thị giác máy tính. “Về cơ bản, nó giống với mô hình mà mọi người sử dụng để tổng hợp hình ảnh trên máy tính. Chúng tôi đang coi mỗi cấu trúc phân tử như một hình ảnh mà mô hình có thể học được. Vì vậy, dự án này đang được xây dựng dựa trên nỗ lực của cộng đồng máy học”, Zhang cho biết.
Trong khi việc điều chỉnh phương pháp tiếp cận thị giác máy tính với hóa học là sự đổi mới quan trọng của DeepBAR, sự giao nhau cũng đặt ra một số thách thức. “Những mô hình này ban đầu được phát triển cho hình ảnh 2D. Nhưng ở đây chúng tôi có protein và phân tử - có cấu trúc 3D. Vì vậy, việc điều chỉnh những phương pháp đó trong trường hợp của chúng tôi là thách thức kỹ thuật lớn nhất mà chúng tôi phải vượt qua”, Ding nói.
Sàng lọc thuốc sẽ nhanh hơn trong tương lai
Trong các thử nghiệm sử dụng các phân tử nhỏ giống như protein, DeepBAR đã tính toán năng lượng tự do liên kết nhanh hơn gần 50 lần so với các phương pháp trước đây. Zhang nói rằng hiệu quả này có nghĩa là “chúng ta thực sự có thể bắt đầu nghĩ đến việc sử dụng kỹ thuật này để thử nghiệm thuốc, đặc biệt là trong bối cảnh COVID. DeepBAR có độ chính xác giống như một tiêu chuẩn vàng, nhưng nó có tốc độ nhanh hơn nhiều”.
Các nhà nghiên cứu nói thêm rằng, ngoài việc sàng lọc thuốc, DeepBAR có thể hỗ trợ phát triển và thiết kế protein vì phương pháp này có thể được sử dụng để tạo mô hình tương tác giữa nhiều loại protein.
Michael Gilson, giáo sư khoa học dược phẩm tại Trường Đại học California tại San Diego, không tham gia vào việc nghiên cứu, cho biết, DeepBAR là “một công trình tính toán thực sự tuyệt vời” với một vài rào cản cần phải giải quyết. Ông nói rằng, “DeepBAR sẽ cần phải được xác thực dựa trên dữ liệu thử nghiệm phức tạp. Điều đó chắc chắn sẽ đặt ra những thách thức bổ sung và nó có thể yêu cầu thêm các giá trị gần đúng hơn”.
Trong tương lai, các nhà nghiên cứu có kế hoạch cải thiện khả năng tính toán của DeepBAR đối với các protein lớn, một nhiệm vụ khả thi nhờ những tiến bộ gần đây trong khoa học máy tính.
Ding nói: “Nghiên cứu này là một ví dụ về việc kết hợp các phương pháp hóa học tính toán truyền thống, được phát triển trong nhiều thập kỷ, với những phát triển mới nhất trong học máy. Vì vậy, chúng tôi đã đạt được điều mà trước đây có thể là không thể”.
Theo https://phys.org/news/2021-03-faster-drug-discovery-machine.html
Bình luận
Tin khác

Ấn tượng Lễ trao giải Cuộc thi vẽ tranh “Bảo vệ môi trường trong học đường – Sân trường mơ ước của em”

“Thắp lửa trái tim” – Lan tỏa yêu thương, gần 700 triệu đồng gửi về vùng cao

Phát động Cuộc thi vẽ tranh Bảo vệ môi trường trong học đường lần thứ II

Gần 750 bác sĩ nhận bằng tốt nghiệp

Văn phòng AOSC phối hợp cùng QUATEST 3 và Vinatest thực hiện chuyên đề giới thiệu ISO 15189
Tin cũ hơn

Trung tâm Đo lường – Nhà máy Z176: Không ngừng hoàn thiện hệ thống quản lý theo tiêu chuẩn ISO/IEC 17025:2017

Gần 1.000 bài viết tham dự cuộc thi “Vượt lên số phận” lần thứ VII

Sắp diễn ra giải bóng đá "Báo chí đồng hành cùng doanh nghiệp"

Lan toả chương trình trồng cây "Đường xanh" đến cộng đồng

Đã có gần 18.000 người tham gia Chiến dịch "Triệu bước chân nhân ái"











